





If you’re preparing to market a medical device in the United States, understanding what is FDA 510(k) clearance is not optional. It is the regulatory pathway that most Class II medical devices must pass through before they can legally reach the market. Yet manufacturers routinely conflate clearance with approval, underestimate documentation demands, or choose predicates that undermine their entire submission. This guide cuts through that confusion, explains the statutory basis of substantial equivalence, and walks you through every critical stage of the FDA 510(k) process from selection to submission.
Navigating Your FDA 510(k) Clearance Journey
- Key Takeaways
- What FDA 510(k) clearance actually means
- The FDA 510(k) submission process, step by step
- Documentation and evidence requirements
- Predicate device selection and evaluation
- Common pitfalls and how to avoid them
- My perspective on what manufacturers consistently get wrong
- How Jjccgroup supports your 510(k) clearance strategy
- FAQ
Key Takeaways
| Point | Details |
|---|---|
| Clearance is not approval | FDA 510(k) clearance means substantial equivalence to a predicate, not a full safety and effectiveness determination. |
| Predicate selection is foundational | Choosing an invalid or mismatched predicate can halt your clearance before substantive review begins. |
| The 90-day clock has caveats | FDA targets a 90-calendar-day decision, but information requests pause the clock and shift your real timeline. |
| Documentation consistency matters | Mismatches between intended use, labeling, and test data are among the most common causes of review delays. |
| Early planning reduces risk | Starting regulatory strategy before design freeze significantly improves submission quality and review speed. |
What FDA 510(k) clearance actually means
The term “FDA 510(k) clearance” refers to the agency’s determination that a new medical device is substantially equivalent to a legally marketed predicate device. The name traces directly to Section 510(k) of the Federal Food, Drug, and Cosmetic Act, which requires device manufacturers to notify FDA at least 90 days before marketing certain devices. That notification became the submission mechanism the industry now calls a 510(k).
Here is where many manufacturers go wrong from the start. FDA clearance is not the same as FDA approval. Approval, which applies to Class III devices through the Premarket Approval pathway (PMA), requires the manufacturer to independently demonstrate that the device is safe and effective. Clearance means something different: the FDA has determined your device is substantially equivalent to a legally marketed predicate. As substantial equivalence is the legal standard underpinning 510(k) clearance, the quality of your predicate comparison drives everything.
Understanding FDA 510(k) in practical terms means recognizing these core concepts:
- Substantial equivalence requires that the new device share the same intended use as the predicate. If technological characteristics differ, those differences must not raise new questions of safety or effectiveness.
- Predicate devices can be pre-amendment devices (marketed before May 28, 1976), devices previously cleared via 510(k), or devices authorized through De Novo classification.
- 510(k) vs PMA is not simply a matter of device class. Some Class II devices may require PMA if they present higher risk. Classification determines the required pathway.
- FDA clearance types include Traditional 510(k), Abbreviated 510(k) for established guidance-based submissions, and Special 510(k) for device modifications by the original submitter.
“Substantial equivalence does not require identical devices. It demands rigorous demonstration that differences do not raise safety or effectiveness concerns.”
This distinction shapes how you frame every document in your submission package.
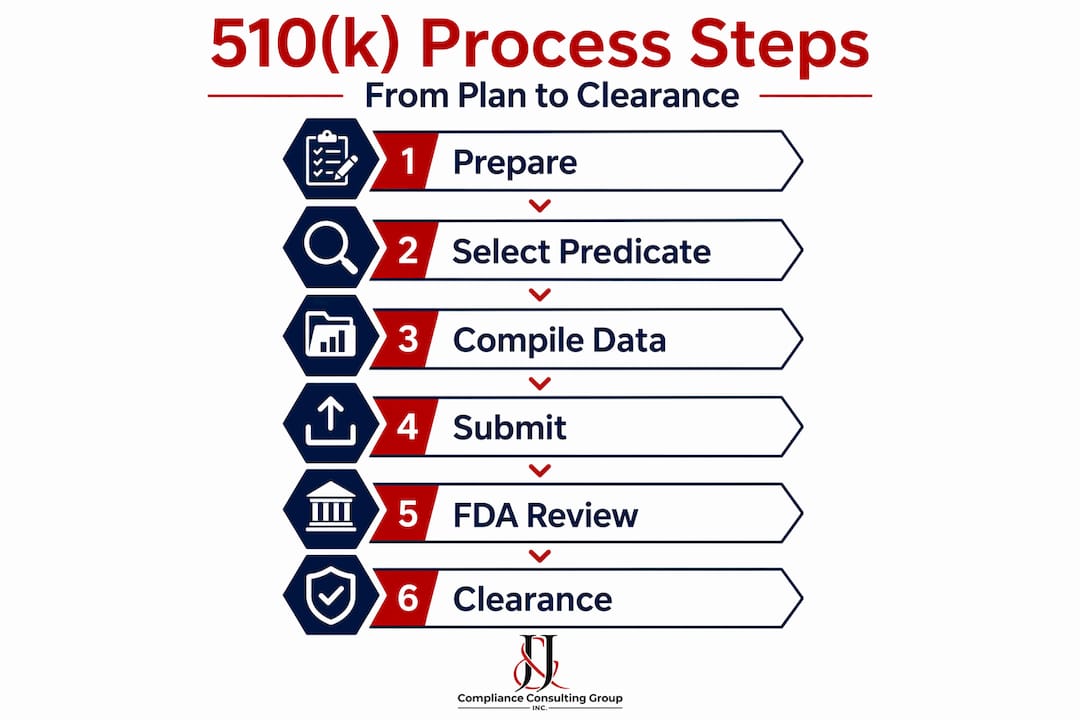
The FDA 510(k) submission process, step by step
How 510(k) works procedurally is something every manufacturer should map out before writing a single page of their submission. The process begins well before the actual filing date, and its outcome depends heavily on preparation quality.
Here is the sequence of events in a standard Traditional 510(k) submission:
- Pre-submission meeting (optional but advisable): Request a Q-Submission to align with FDA on your intended use statement, predicate choice, and any testing questions before committing to a full submission package.
- Prepare the submission package: Assemble all required sections, including the cover letter, device description, substantial equivalence discussion, performance testing, and labeling.
- Submit to the FDA: File electronically through the eSTAR system. FDA conducts an initial administrative check.
- Acceptance review: FDA has 15 business days to determine whether the submission meets the Refuse to Accept (RTA) checklist. Incomplete submissions are rejected and must be resubmitted.
- Substantive review: Once accepted, the 90-day review clock begins. The assigned reviewer evaluates your substantial equivalence argument and supporting data.
- Additional Information (AI) requests: If FDA needs clarification or more data, they issue an AI request. The clock pauses until you respond, which can significantly extend your real-world timeline.
- Decision: FDA issues a clearance order (substantially equivalent), a Not Substantially Equivalent (NSE) determination, or requests additional actions.
Pro Tip: Build at least 30 to 60 additional days into your project plan beyond the nominal 90-day review window. Administrative screening and AI requests routinely push real decision dates well past the calendar target, and teams that plan for the benchmark alone consistently miss internal launch milestones.
The difference between a smooth review and a prolonged one often comes down to whether you anticipated FDA’s questions before they asked them.

Documentation and evidence requirements
Meeting 510(k) clearance requirements on paper is not enough. Your documentation must form a coherent, traceable argument from intended use through design controls to verified performance. FDA reviewers are reading your submission as a story. If the plot has gaps, they will ask for rewrites.
The core documentation components to get right include:
- Intended use and indications for use: These two statements are related but not identical. Your intended use defines what the device does. Indications for use describe the specific conditions or patient populations. Every other document in your submission should align with these definitions without exception.
- Technological characteristics comparison: Identify where your device differs from the predicate and provide objective data showing those differences do not compromise safety or effectiveness. This is the heart of your substantial equivalence argument.
- Performance testing: FDA expects bench testing against recognized standards, biological safety testing per ISO 10993 for patient-contacting devices, and software documentation per FDA guidance if your device contains software. For implantable devices, robust preclinical testing is expected even when clinical trials are not required.
- Biocompatibility data: Any device with patient contact requires a biocompatibility evaluation. FDA scrutinizes this section closely for completeness of endpoints evaluated.
- Human factors documentation: For devices with user interfaces, FDA increasingly expects formative and summative usability testing reports.
- Traceability: Manufacturers must maintain traceability matrices linking design requirements to design outputs and risk controls to verification activities. This demonstrates a coherent safety and effectiveness story that FDA can follow without guessing.
Pro Tip: Create a cross-reference matrix before finalizing your 510(k) submission. Every claim in your substantial equivalence discussion should point to a specific test report, standard, or risk control document. Reviewers who can find the evidence quickly are far less likely to issue AI requests.
Understanding the 510(k) summary document requirements is also part of building a complete package. That summary becomes publicly available upon clearance, so it must accurately reflect your cleared claims.
Predicate device selection and evaluation
Selecting your predicate device is the single most consequential decision in your 510(k) strategy. Everything that follows, including your substantial equivalence argument, your performance testing scope, and your labeling, flows from this choice.
A valid predicate must be legally marketed in the United States and must share the same intended use as your device. Predicate devices can fall into three categories: pre-amendment devices marketed before 1976, devices with 510(k) clearance, and devices with De Novo authorization. PMA-approved devices cannot serve as predicates.
The table below outlines the key evaluation criteria for predicate selection:
| Evaluation Criterion | What FDA Looks For |
|---|---|
| Intended use alignment | Exact match or narrower scope in the new device |
| Technological characteristics | Similarity or justified differences with supporting data |
| Legal market status | Confirmed clearance or pre-amendment standing |
| Risk profile | No new risks introduced by technological differences |
| Labeling consistency | Predicate’s cleared indications support your claims |
FDA reviewers assess submissions through a predicate validity lens. Using an unsuitable predicate can trigger an RTA finding or an NSE determination, both of which cost months of rework. Search FDA’s 510(k) database (accessible at fda.gov) to review prior clearances, including the cleared indications and device descriptions. Read the predicate’s 510(k) summary carefully. If the predicate itself was cleared based on a narrow indication, assuming it covers your broader use is a risk that FDA will surface during review.
Some manufacturers use a split predicate strategy, referencing one device for intended use and a second for technological characteristics. This is permitted but requires careful justification and is sometimes more difficult to execute convincingly.
Common pitfalls and how to avoid them
The FDA 510(k) submission process rewards preparation and penalizes assumptions. Across submissions, the patterns that stall clearance are remarkably consistent.
- Inconsistent intended use statements: If your cover letter, device description, labeling, and substantial equivalence discussion each describe your device’s purpose in slightly different terms, FDA will flag the inconsistency. Poor cross-document consistency is one of the most reliable predictors of an AI request.
- Gaps in the performance testing rationale: Failing to justify why certain tests were not performed, or citing standards that do not fully apply to your device type, invites questions. Explicitly address any deviations from FDA guidance documents or recognized standards.
- Weak risk analysis linkage: Your risk management file should not be a standalone document. Risk controls should connect directly to design outputs and verification evidence throughout the submission.
- Software and cybersecurity gaps: If your device contains software, FDA expects documentation aligned with their Software as a Medical Device (SaMD) guidance and, increasingly, cybersecurity documentation per the 2023 Cybersecurity Guidance. Omitting this entirely for a connected device is a significant gap.
- Reactive rather than proactive planning: Many manufacturers engage regulatory support after design is frozen, when making changes is expensive. Starting earlier, including using a pre-submission meeting to align with FDA, reduces the risk of late-stage surprises.
Submissions that succeed consistently share one quality: clear, evidence-linked narratives that connect intended use, risk controls, and objective performance data into a single coherent argument.
My perspective on what manufacturers consistently get wrong
I’ve worked alongside manufacturers at every stage of the 510(k) process, and the pattern I see most often is not a lack of technical rigor. It is a failure to treat the submission as a regulatory argument rather than a document-filing exercise.
Teams spend months generating test data and then lose weeks in review because no one asked, “Does this test actually address the safety question FDA will ask about our predicate differences?” The data exists. The link to the argument doesn’t.
What I’ve found works is treating substantial equivalence as a story with a logical structure. Start with intended use. Map every design decision, risk control, and test result back to that use. When a reviewer can follow that thread from page one to the final performance report without losing it, you get decisions, not questions.
The other lesson I’ve learned: the manufacturers who treat the 90-day review window as their planning horizon are the ones who are surprised by AI requests. Build a realistic schedule that accounts for the RTA screening, likely AI interactions, and your own internal response time. That is not pessimism. That is how this process actually works.
If you are early in development, get expert consulting support before design freeze. That single decision will save more time and money than any other investment in your submission strategy.
— Mike
How Jjccgroup supports your 510(k) clearance strategy
Preparing a 510(k) submission without deep regulatory experience is a high-stakes gamble. Every gap in documentation, every misaligned intended use statement, and every poorly justified predicate comparison costs you time and extends your path to market.

Jjccgroup brings over 30 years of regulatory consulting experience to manufacturers navigating the FDA 510(k) process. From predicate strategy and pre-submission meeting preparation to full submission package review and post-submission support, Jjccgroup provides the structured expertise that reduces your review risk. Their regulatory approval consulting services are designed specifically for medical device manufacturers who need more than generic guidance. They need a partner who understands FDA expectations at the reviewer level and can help you build a submission that answers questions before they are asked. If your device is approaching design freeze or your submission is already drafted, this is the right time to engage expert support.
FAQ
What is FDA 510(k) clearance in simple terms?
FDA 510(k) clearance is a regulatory determination that your new medical device is substantially equivalent to a legally marketed predicate device, allowing you to market it in the United States. It is not the same as FDA approval, which requires independent proof of safety and effectiveness.
How long does the FDA 510(k) review take?
FDA targets a 90-calendar-day decision from the acceptance date, but the clock pauses during Additional Information requests. Real-world timelines often run longer when you include administrative screening and response periods.
What is a predicate device in a 510(k) submission?
A predicate device is a legally marketed device that your new device is compared against to establish substantial equivalence. It must share the same intended use and be either a pre-amendment device, a 510(k)-cleared device, or a De Novo authorized device.
Do I need clinical trials for a 510(k) submission?
Clinical trials are not always required. Most 510(k)s rely on nonclinical performance data and biological safety testing. However, implantable or higher-risk devices may require clinical data to support the substantial equivalence argument.
What is the difference between 510(k) and PMA?
The 510(k) pathway requires demonstrating substantial equivalence to a predicate device and applies primarily to Class II devices. PMA is required for Class III devices and demands independent clinical evidence that the device is safe and effective, making it a significantly more demanding and time-intensive process.