





Medical device import compliance is the legally mandated practice of ensuring imported devices meet all applicable regulatory standards required by jurisdictions like the FDA in the U.S., the EU MDR framework, and Health Canada before and after market entry. Without this compliance, devices cannot legally enter or remain in any of these markets. For regulatory compliance officers and import managers, understanding why medical device imports need compliance is not optional background knowledge. It is the operational foundation that determines whether your products reach patients or get detained at the port.
Why medical device imports need compliance across major markets
The core reason why compliance matters for imports is straightforward: every major regulatory authority treats the import event as a formal compliance checkpoint, not a formality. FDA enforces import regulations at the point of entry, and noncompliant devices can be detained immediately. The FDA does not recognize other countries’ regulatory approvals as substitutes for its own requirements. That means a device cleared by Health Canada or CE-marked under EU MDR still requires independent FDA compliance before it enters U.S. commerce.
The EU MDR framework under Regulation (EU) 2017/745 places legal obligations on importers to verify CE conformity independently before placing any device on the European market. This is not a passive role. EU importers act as gatekeepers with their own verification duties, separate from the manufacturer’s technical file. In Canada, importers must hold an establishment license and confirm the device manufacturer holds a valid license for Class II through IV devices. Failing to confirm upstream licensing stalls imports even when all technical documentation is complete.

The table below summarizes the core import compliance requirements across the three major jurisdictions.
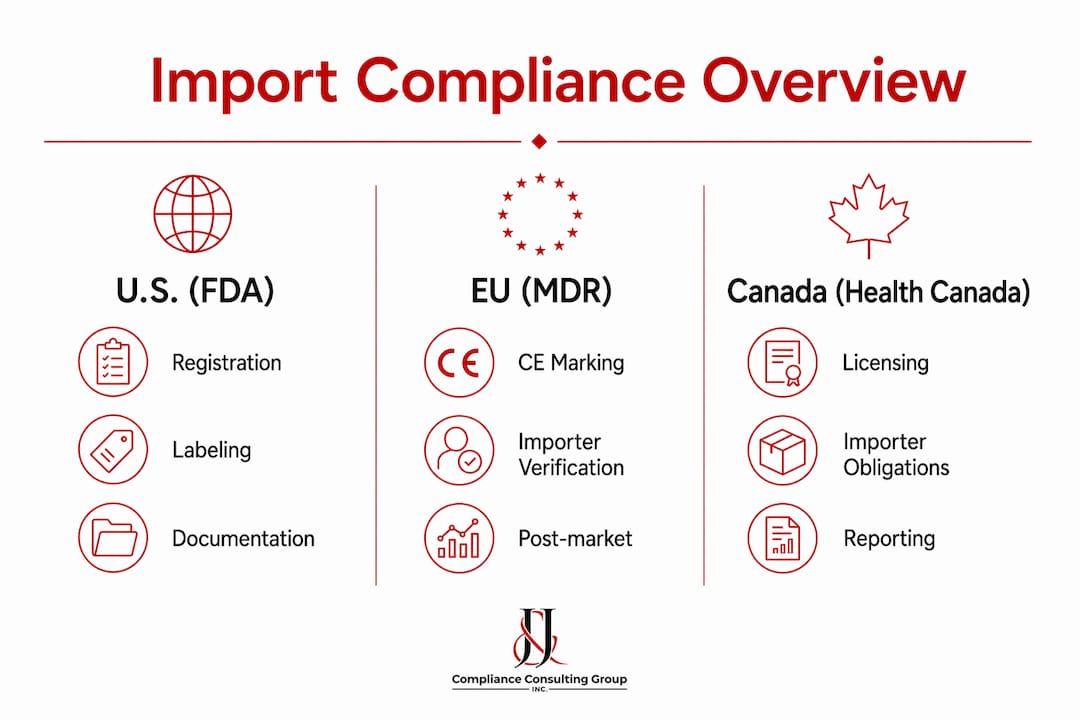
| Requirement | U.S. (FDA) | EU (MDR) | Canada (Health Canada) |
|---|---|---|---|
| Establishment registration | Required for importers and manufacturers | Not applicable; CE marking required | Establishment license mandatory |
| Device listing or approval | Premarket clearance or approval (510(k), PMA) | CE conformity under EU MDR | Device license for Class II–IV |
| Labeling compliance | FDA-specific labeling standards | EU language and labeling requirements | Bilingual labeling (English/French) |
| Post-market reporting | MDR adverse event reporting | Cooperation with recalls and vigilance | Incident reporting to Health Canada |
| Importer documentation | Entry documentation, importer records | Conformity verification records | Import notification and license verification |
These requirements share a common logic: the importer bears legal responsibility for what enters the market, not just the manufacturer. That shared logic is why compliance for medical device safety cannot be delegated entirely to the foreign manufacturer.
How compliance impacts market access and supply chain reliability
Non-compliance does not produce a warning letter and a second chance at the port. Failure to maintain compliance causes shipment detention, financial loss, supply chain disruptions, and possible market withdrawal. For import managers, a single detained shipment can cascade into back-ordered products, broken hospital contracts, and regulatory scrutiny that follows the company for years.
The operational consequences are specific and worth naming:
- Shipment detention: FDA or Customs and Border Protection can place a device on import alert, requiring the importer to prove compliance before release. This process can take weeks.
- Refusal of admission: Devices that cannot demonstrate compliance are refused entry and must be re-exported or destroyed at the importer’s expense.
- Recalls and market withdrawal: Devices that enter the market without proper premarket authorization or with labeling deficiencies face mandatory recalls under FDA authority.
- Financial penalties: Regulatory violations under the Federal Food, Drug, and Cosmetic Act carry civil monetary penalties and, in serious cases, criminal liability.
- Reputational damage: Enforcement actions are publicly posted on FDA’s import alert database, visible to customers, partners, and competitors.
Pre-entry compliance reconciliation is the most effective operational strategy to prevent these outcomes. This means aligning device identity data, importer records, labeling content, and premarket status before the shipment leaves the manufacturer’s facility. Import compliance must integrate with supply chain workflows, not run parallel to them.
Pro Tip: Build a pre-shipment compliance checklist that cross-references FDA establishment registration numbers, device listing codes, and labeling version control against each purchase order. Catching a labeling mismatch before the shipment departs costs hours. Catching it at the port costs weeks.
What post-import compliance responsibilities do importers hold?
Compliance does not end when the device clears customs. FDA frames compliance as a lifecycle safety and effectiveness obligation monitored through total product lifecycle activities, including inspections, post-market surveillance, and quality management system requirements. Import managers who treat compliance as a one-time entry event expose their organizations to ongoing enforcement risk.
Post-import obligations vary by jurisdiction but follow a consistent pattern of reporting, record-keeping, and cooperation with regulatory authorities.
- U.S. MDR reporting: Initial importers must comply with Medical Device Reporting requirements, forwarding adverse event reports and maintaining MDR event files. The “initial importer” designation under FDA regulations is not just a customs classification. It triggers specific post-market obligations that attach permanently to the entity named at entry.
- Complaint handling: FDA requires importers to maintain complaint files and forward complaints to manufacturers in a documented, traceable manner.
- EU importer duties: EU importers must keep records of complaints, recalls, and devices under their responsibility, cooperate with authorities on corrective actions, and verify translation and labeling accuracy as part of a quality management system. Critically, EU MDR importers can create independent compliance breaches even when the manufacturer’s technical file is accurate, if the importer’s own verification records are incomplete.
- Canada post-import duties: Health Canada requires importers to report incidents involving serious injury or death, update labeling when required, and maintain their establishment license in good standing throughout the device’s market life.
- Quality management alignment: Maintaining a quality management system aligned to ISO 13485 is the most reliable mechanism for meeting post-import obligations across all three jurisdictions simultaneously.
Pro Tip: Assign a named regulatory contact within your organization for each market where you hold importer status. Regulatory agencies expect a responsive, knowledgeable point of contact. An unanswered inquiry from FDA or a notified body during a post-market review escalates faster than most compliance officers anticipate.
Common compliance challenges and best practices for import managers
The most frequent compliance failures in medical device imports are not caused by ignorance of the regulations. They result from misaligned contracts, documentation gaps, and organizational confusion about who holds the importer role. Understanding these pitfalls is the first step toward preventing them.
Misidentifying the importer of record is the single most consequential error. Under FDA rules, importer identity dictates post-market obligations like MDR event file maintenance and complaint forwarding. If your contracts assign customs brokerage to a third party without clarifying FDA importer status, your organization may unknowingly hold regulatory obligations it is not operationally prepared to fulfill.
Documentation discrepancies between the manufacturer’s technical file, the importer’s entry records, and the device label are a leading cause of port holds. FDA treats the import event as a compliance checkpoint combining device identity, importer data, labeling, premarket status, and regulatory classification codes. Misalignment across any of these data points escalates to detention.
Labeling errors are particularly costly because they affect both entry compliance and post-market standing. Incorrect indications for use, missing required statements, or labeling not translated into required languages (French in Canada, EU member state languages under MDR) trigger refusal or recall.
The following best practices address these challenges directly:
- Clarify importer of record in every contract. Specify which entity holds FDA initial importer status, EU MDR importer status, or Canadian establishment license obligations in writing before the first shipment.
- Conduct pre-import compliance reviews. Before each new product or new supplier relationship, verify premarket authorization status, labeling compliance, and registration currency in all target markets.
- Align your quality management system to ISO 13485. The ISO 13485 standard provides the procedural framework for complaint handling, record retention, and corrective action that satisfies FDA, EU MDR, and Health Canada simultaneously.
- Maintain a regulatory change log. Medical device import standards evolve. EU MDR transition deadlines, FDA QMSR updates, and Health Canada guidance revisions require active monitoring, not annual reviews.
- Engage regulatory consulting for complex classifications. Devices that span multiple regulatory categories or involve novel technologies benefit from expert classification review before import, not after a detention notice.
- Train your supply chain team on compliance roles. Logistics personnel who do not understand the difference between a customs broker and an FDA initial importer create organizational risk that no compliance checklist can fully mitigate.
Pro Tip: When onboarding a new foreign manufacturer, request their FDA establishment registration number, EU authorized representative contact, and Canadian device license number before signing the supply agreement. Verifying these upstream credentials takes 30 minutes. Resolving a detention caused by an expired manufacturer license takes months.
Key takeaways
Medical device import compliance is a continuous legal obligation across the U.S., EU, and Canada, covering entry verification, post-market reporting, and quality management throughout the device lifecycle.
| Point | Details |
|---|---|
| Compliance is mandatory at entry | FDA, EU MDR, and Health Canada each require independent compliance verification before market placement. |
| Importer role carries legal weight | The entity named as importer holds specific post-market obligations including MDR reporting and complaint handling. |
| Non-compliance disrupts supply chains | Shipment detention, refusal of admission, and market withdrawal are direct consequences of import compliance failures. |
| Post-import duties extend the lifecycle | Adverse event reporting, record-keeping, and cooperation with recalls are ongoing importer responsibilities in all three jurisdictions. |
| Best practices prevent most failures | Pre-import reviews, ISO 13485 alignment, and clear contract language resolve the majority of common compliance gaps. |
What I’ve learned from years of watching import compliance fail at the wrong moment
The pattern I see most often is not a company that ignored compliance. It is a company that believed compliance was someone else’s job. The manufacturer assumed the importer handled FDA registration. The importer assumed the customs broker managed labeling verification. The customs broker assumed the manufacturer’s documentation was current. Nobody checked. The shipment arrived at a U.S. port with an expired establishment registration, and three weeks of detention followed.
What strikes me about this scenario is how preventable it is. The medical device import regulations are specific about who owns each obligation. The problem is organizational, not regulatory. Companies that build compliance accountability into their supplier contracts and internal workflows almost never face detention. Companies that treat compliance as a documentation exercise rather than an operational discipline face it repeatedly.
The other insight worth sharing is that proactive quality systems do more than prevent enforcement. They accelerate market access. When your establishment registration is current, your labeling is pre-reviewed, and your MDR reporting procedures are documented, entry moves faster. Regulators at every port and border agency have discretion in how deeply they examine a shipment. A well-documented importer with a clean compliance history gets less scrutiny, not more.
The regulatory expectations in 2026 are more specific than they were five years ago. EU MDR is fully enforced. FDA’s Quality Management System Regulation updates are in effect. Health Canada continues tightening establishment licensing oversight. Import managers who embed compliance into their supply chain workflows now will spend far less time in reactive mode later.
— Mike
How Jjccgroup supports medical device import compliance

Jjccgroup brings over 30 years of regulatory expertise to medical device import compliance across the U.S., EU, and Canada. Whether you need FDA establishment registration support, EU MDR importer obligation reviews, or Canadian establishment licensing guidance, Jjccgroup provides the regulatory approval consulting that keeps your supply chain moving and your compliance record clean. Services include labeling review, importer role management, audit preparation, and quality management system alignment to ISO 13485. If your organization is managing complex import classifications or preparing for a new market entry, working with an experienced consulting partner reduces the risk of costly enforcement actions and accelerates your path to market.
FAQ
What does medical device import compliance require?
Medical device import compliance requires that devices meet all applicable regulatory standards of the destination market before entry, including premarket authorization, labeling compliance, establishment registration, and post-market reporting obligations.
Why can’t FDA accept CE marking or Health Canada approval instead?
FDA does not recognize other countries’ regulatory approvals as substitutes for its own requirements. Each jurisdiction maintains independent compliance standards, and importers must satisfy each authority separately.
What happens if a medical device fails import compliance?
Non-compliant devices face shipment detention, refusal of admission, or mandatory recall. Financial penalties and public enforcement listings on FDA’s import alert database are additional consequences that affect future shipments.
Who is responsible for compliance when importing medical devices?
The entity designated as the importer of record holds primary compliance responsibility, including post-market MDR reporting, complaint handling, and record retention. This designation must be explicitly defined in supplier and logistics contracts.
How does ISO 13485 support import compliance?
ISO 13485 provides the quality management framework that satisfies complaint handling, corrective action, and record-keeping requirements across FDA, EU MDR, and Health Canada simultaneously, making it the most efficient single standard for multi-market importers.