





If you’ve ever braced for an FDA inspection wondering whether you’re truly prepared, you’re not alone. Understanding why FDA audits manufacturers goes far beyond knowing that regulations exist. These inspections, formally called FDA regulatory inspections, are the agency’s primary mechanism for verifying that your products are safe, effective, and consistently manufactured to established quality standards. The FDA’s mandate spans pharmaceuticals, medical devices, food and beverage, dietary supplements, and tobacco. That means millions of patients and consumers depend on the outcomes of these audits every single year.
Table of Contents
- Key Takeaways
- Why FDA audits manufacturers: the foundational answer
- How FDA selects facilities for inspection
- What to expect during an FDA inspection today
- Why these inspections matter beyond a checkbox
- Practical steps to prepare for your next FDA inspection
- My perspective on what FDA audits really reveal
- How Jjccgroup helps manufacturers stay ahead of FDA inspections
- FAQ
Key Takeaways
| Point | Details |
|---|---|
| FDA audits verify real compliance | Inspections confirm your quality systems function as intended, not just on paper. |
| Risk scores determine audit frequency | Prior inspection history, recalls, and complaint data all influence how often FDA visits your facility. |
| QMSR changes what auditors examine | The 2026 Quality Management System Regulation shifts focus to integrated quality processes, not isolated subsystems. |
| ISO certification does not replace FDA inspection | Holding an ISO 13485 certificate does not exempt you from FDA regulatory oversight or inspections. |
| Proactive preparation reduces enforcement risk | Internal audits, mock inspections, and documented CAPA processes significantly reduce your risk of receiving serious citations. |
Why FDA audits manufacturers: the foundational answer
The FDA conducts inspections to verify manufacturer compliance with current Good Manufacturing Practice (cGMP) requirements, protecting product safety, efficacy, and data integrity. That is the official answer. But the fuller picture involves understanding what these audits actually examine and how they differ from other forms of quality oversight.
FDA regulatory inspections are not the same as ISO certification audits or third-party quality reviews. ISO 13485 certification is a market-facing credential. An FDA inspection is a legal enforcement activity conducted under statutory authority. The FDA can take regulatory action based on inspection findings. An ISO registrar cannot. That distinction matters enormously when you are preparing your quality team.
There are four primary types of FDA inspections manufacturers encounter:
- Routine surveillance inspections: Conducted on a scheduled basis to monitor ongoing cGMP compliance across regulated industries.
- Pre-approval inspections (PAI): Triggered when a manufacturer submits a new product application, confirming the facility can produce the product as described in the submission.
- For-cause inspections: Initiated in response to specific complaints, adverse events, recalls, or other red flags in the agency’s surveillance data.
- Unannounced inspections: Conducted without prior notice, giving FDA investigators an unfiltered view of your day-to-day operations.
Understanding which type of inspection you may be facing changes how you need to prepare and respond.
How FDA selects facilities for inspection
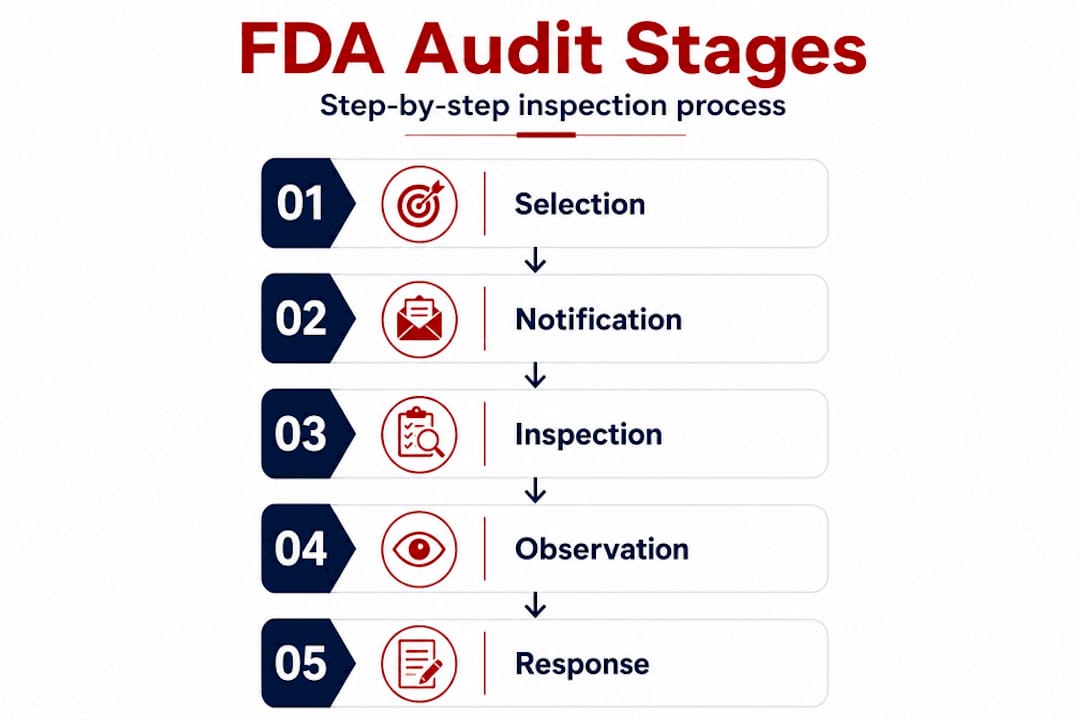
FDA does not inspect every facility every year. Resources are finite, so the agency uses a risk-based Site Selection Model (SSM) to prioritize which manufacturing sites receive attention and when. The model evaluates multiple factors to generate a risk score for each facility.
The key factors include:
- Prior inspection outcomes: A facility with a history of Official Action Indicated (OAI) classifications will rank higher in risk than one with consistent Voluntary Action Indicated (VAI) or No Action Indicated (NAI) results.
- Time since last inspection: The longer the gap since your last FDA visit, the higher your priority score generally becomes.
- Product risk profile: Higher-risk products such as sterile injectables or Class III medical devices receive more frequent scrutiny than lower-risk product categories.
- Compliance history and geographic data: Regional compliance trends factor into FDA’s resource allocation decisions.
- Post-market surveillance signals: Reports of product recalls, adverse events, Field Alert Reports, MedWatch submissions, and quality complaints all increase a facility’s inspection priority score.
This last point deserves emphasis. A single product recall does not just cause short-term operational disruption. It can place your facility under elevated FDA scrutiny for years. That is a concrete reason why proactive quality management matters beyond simple compliance.
Pro Tip: Monitor your own post-market surveillance data the way FDA does. If your complaint rates, adverse event reports, or field corrections are trending upward, expect that FDA’s risk score for your facility is trending upward too. Address systemic issues before an investigator flags them.
What to expect during an FDA inspection today
The nature of FDA inspections has shifted meaningfully in 2026. The transition to the Quality Management System Regulation (QMSR) has replaced the legacy Quality System Inspection Technique (QSIT) with a process-based inspection model under Compliance Program 7382.850. This shift has real implications for what FDA investigators examine when they walk through your door.

Under the legacy QSIT approach, investigators evaluated discrete quality subsystems independently. Under the new process-based model, they assess how your quality elements function together as an integrated system. That means your management review process, internal audit program, corrective and preventive action (CAPA) system, risk management integration, and supplier controls are all evaluated in relation to each other, not in isolation.
Here is a practical comparison of what that looks like:
| Focus area | Legacy QSIT approach | 2026 QMSR process-based approach |
|---|---|---|
| Management review | Reviewed as a separate subsystem | Evaluated as evidence of quality system maturity |
| CAPA | Assessed for documentation completeness | Assessed for systemic root cause analysis and risk linkage |
| Risk management | Minimal direct inspection focus | Integrated into all quality system elements |
| Supplier controls | Checked for basic documentation | Evaluated as part of the overall supply chain risk picture |
| Internal audits | Existence and frequency reviewed | Depth, findings, and follow-through examined |
A critical misconception to address here: ISO 13485 certification does not exempt medical device manufacturers from FDA inspections. The FDA explicitly states that certificates of conformance to ISO 13485 do not replace inspections focused on FDA-specific regulatory compliance. Many manufacturers invest significantly in ISO certification while their FDA-specific documentation gaps go unaddressed. Those gaps tend to surface quickly under a process-based inspection.
Pro Tip: Review your CAPA records before any inspection and ask whether each entry demonstrates a systemic root cause analysis. If your CAPA documentation mostly describes what happened rather than why it happened and what changed systematically, that is a red flag worth addressing immediately.
Why these inspections matter beyond a checkbox
The importance of FDA audits extends well past regulatory compliance. FDA emphasizes that quality cannot simply be inspected into products. Quality must be built into design, development, and manufacturing processes, and it must be reflected in a genuine quality culture throughout your organization.
That philosophy shapes how audits are conducted and what their findings reveal. When an FDA investigator issues Form 483 observations at the close of an inspection, those observations represent identified departures from cGMP requirements. The agency then tracks inspection follow-ups to confirm that manufacturers take appropriate corrective actions, particularly for significant violations that carry public health risk.
The escalation path matters. Form 483 observations that go unaddressed or receive inadequate responses can lead to Warning Letters, consent decrees, import alerts, or mandatory recalls. Each step in that escalation ladder carries costs that dwarf the investment required for proactive quality improvement.
“The most important thing FDA audits do is hold manufacturers accountable to the same standards their customers and patients are counting on them to meet. The audit itself is not the threat. The quality gap it reveals is.”
Beyond enforcement, FDA inspections serve as external validation of whether your quality management system is functioning as designed. That kind of objective assessment is difficult to replicate internally. Many manufacturers who engage honestly with audit findings report that the process exposed systemic issues they had not recognized or prioritized, leading to meaningful improvements in product quality and operational reliability.
FDA inspection priorities remain consistent regardless of broader agency leadership changes. Manufacturers who treat compliance as a continuous commitment rather than a pre-inspection sprint consistently fare better across all inspection types.
Practical steps to prepare for your next FDA inspection
Preparation is not a last-minute exercise. The manufacturers who navigate inspections most confidently are those who treat audit readiness as a built-in feature of their quality management system, not a reactive project triggered by receiving an investigator’s notice. Here is a structured approach to getting there:
-
Align your quality system with QMSR requirements. The transition from legacy Quality System Regulation (QSR) to QMSR demands a shift to integrated process thinking rather than document-centric compliance. Map how your CAPA system, risk management files, and supplier quality controls interact and support each other.
-
Maintain current Risk Management Files. FDA investigators increasingly expect to see documented risk management integrated across product lifecycle stages, not confined to a standalone ISO 14971 exercise. Your risk files should reference design, process, and post-market data.
-
Respond to Form 483 observations with depth. If you receive observations, successful Form 483 responses require demonstrating systemic, risk-based root cause analysis that prioritizes patient safety over surface-level explanation. FDA’s 2026 draft guidance on this topic is explicit: describe what changed in your systems, not just what you reviewed.
-
Conduct rigorous internal audits. Internal audits that only confirm existing procedures are compliant with existing procedures provide limited value. Design your internal audit program to stress-test your processes the way an FDA investigator would. Use ISO 13485 internal audit frameworks as a baseline, but push deeper into process integration and risk linkage.
-
Invest in mock inspections. A professional mock FDA inspection conducted by experienced regulatory consultants surfaces documentation gaps, process weaknesses, and personnel readiness issues before an actual investigator finds them. The investment consistently delivers returns far exceeding the cost of reactive corrective action.
Pro Tip: Designate a trained back-room team for inspections. Your front-room team handles the investigator directly. Your back-room team retrieves documents, checks responses for accuracy, and tracks every request in real time. This structure prevents the reactive scrambling that leads to inconsistent or inaccurate answers during an inspection.
My perspective on what FDA audits really reveal
I have worked alongside manufacturers across pharmaceuticals, medical devices, and food production who shared the same initial reaction to an upcoming FDA inspection: dread. And I get it. The stakes are real, the preparation is demanding, and the uncertainty is uncomfortable.
But in my experience, that framing gets it exactly backwards. FDA inspections are not primarily about catching you doing something wrong. They are about confirming that your quality systems actually protect the people using your products. When manufacturers internalize that distinction, their entire approach to compliance shifts. It stops being about passing an audit and starts being about building something worth auditing.
What I have found consistently is that the manufacturers who struggle most with FDA inspections are those whose quality systems exist on paper but not in practice. They have the SOPs, the CAPA records, and the training logs. What they lack is a quality culture where those tools are actually used to drive decisions. FDA investigators are trained to spot exactly that gap.
The transition to QMSR has accelerated this dynamic. A process-based inspection approach rewards manufacturers whose quality systems genuinely function as integrated systems. It exposes those whose compliance infrastructure is more ceremonial than operational. That is not a criticism. It is an opportunity to build something that works.
My honest advice: treat your next FDA inspection as the most rigorous external quality review your organization will receive this year. Then prepare for it with the same seriousness.
— Mike
How Jjccgroup helps manufacturers stay ahead of FDA inspections
Preparing for an FDA inspection is not something most manufacturers need to do alone. Jjccgroup brings over 30 years of regulatory consulting experience to pharmaceutical, medical device, food and beverage, dietary supplement, and tobacco manufacturers navigating exactly these challenges. Whether you need help interpreting 2026 QMSR requirements, responding to Form 483 observations, or building a mock inspection program from scratch, the team provides targeted, practical support.

Jjccgroup’s approach to regulatory approval consulting combines deep FDA compliance expertise with hands-on guidance tailored to your product category and operational reality. Clients consistently report that working with Jjccgroup turns the inspection process from a source of anxiety into a confirmation of the quality systems they have built. If your team is ready to move from reactive compliance to genuine audit confidence, explore the full range of FDA compliance services available or connect with the Jjccgroup team directly.
FAQ
Why does the FDA audit manufacturers in the first place?
The FDA conducts inspections to verify that manufacturers comply with cGMP requirements, confirming that products reaching consumers are safe, effective, and consistently produced to quality standards.
What triggers a for-cause FDA inspection?
Product recalls, adverse event reports, consumer complaints, and Field Alert Reports can all increase a facility’s risk score and trigger a targeted for-cause FDA inspection outside the regular surveillance cycle.
Does ISO 13485 certification eliminate the need for an FDA audit?
No. The FDA explicitly states that ISO 13485 certificates of conformance do not replace FDA inspections, which assess compliance with FDA-specific regulatory requirements rather than ISO standards.
What happens after an FDA investigator issues Form 483 observations?
Manufacturers must provide a written response demonstrating risk-based root cause analysis and corrective actions. Inadequate responses can escalate to Warning Letters, consent decrees, or import alerts.
How often does the FDA inspect a manufacturing facility?
Inspection frequency depends on the facility’s risk score under the FDA’s Site Selection Model, which considers factors including prior inspection outcomes, product risk level, and post-market surveillance signals such as complaints and recalls.
Recommended
- FDA Product Compliance: A Step-by-Step Guide – J&J Consulting Group- FDA Regulatory Compliance
- FDA Regulatory Compliance Consulting: A Guide – J&J Consulting Group- FDA Regulatory Compliance
- FDA Audit Preparation Services & Compliance Experts
- Ace Your Audit: Expert FDA Mock Inspection – J&J Consulting Group- FDA Regulatory Compliance